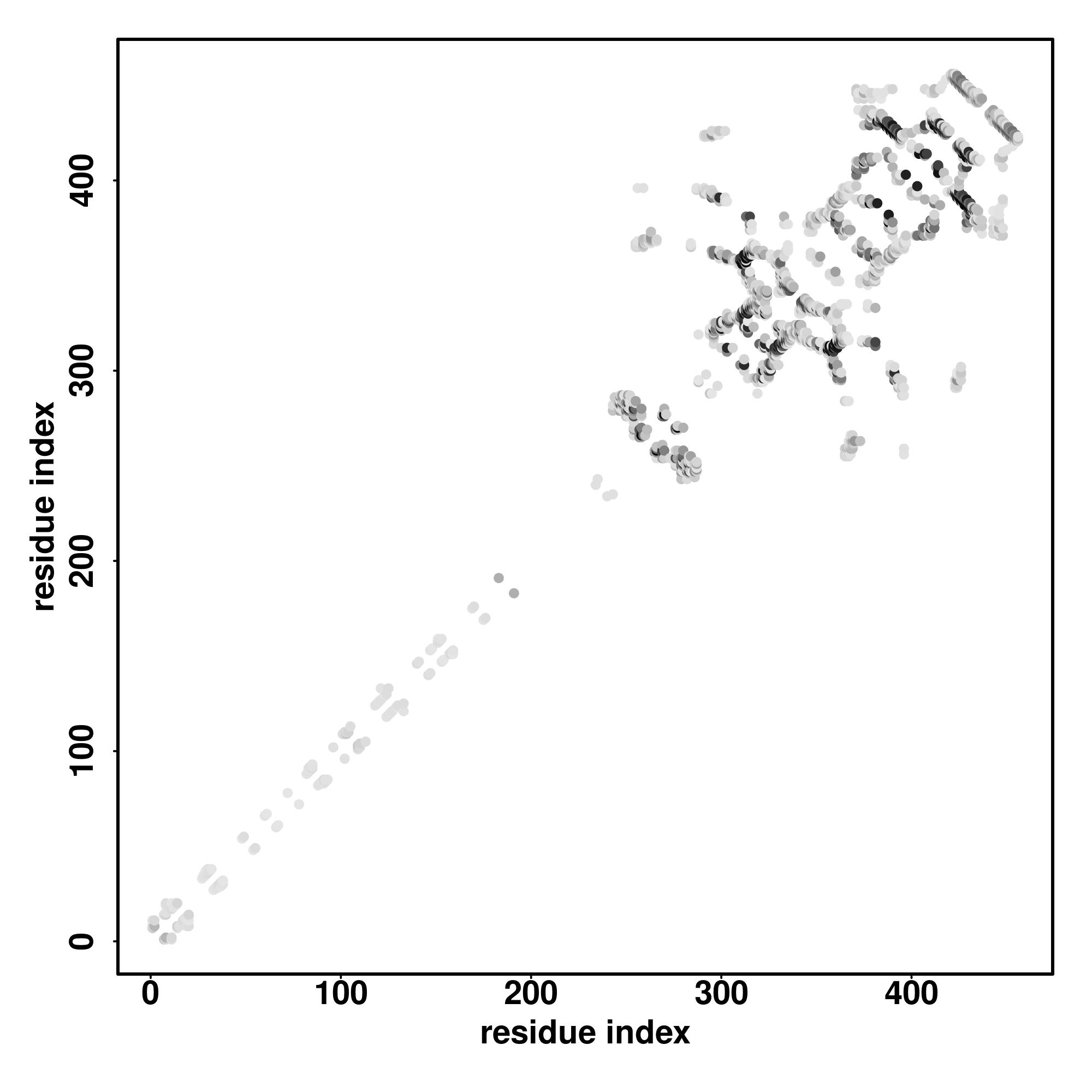
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200
| | | | | | | | | | |
|---|
| SS
Seq | CCCCCHHHHHCCCCCCCCCCCCCCCCCCCCCCCCHHHHHHHHHHHHHHHCCCCCCCCCCCCCCCCCHHHHHHCCCCCCCCHHHCCCCCCHHHCCCCCCCCCCCCCHHHHHHHHHHHHHCCCCCCCCCCHHHHHHHHCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCHHHHHCCCCCCCCCCCCCCCCCCCCCCCCC
MFEEPEWAEAAPVAAGLGPVISRPPPAASSQNKGSKRRQLLATLRALEAASLSQHPPSLCISDSEEEEEERKKKCPKKASFASASAEVGKKGKKKCQKQGPPCSDSEEEVERKKKCHKQALVGSDSAEDEKRKRKCQKHAPINSAQHLDNVDQTGPKAWKGSTTNDPPKQSPGSTSPKPPHTLSRKQWRNRQKNKRRCKNKFQPPQVPDQAPAE |
|---|
| 1 | 3apgA | 0.07 | 0.07 | 2.77 | 0.36 | CEthreader | | PNVVYNQGYINLRSGFPPGYLSFEAAEKAKFNLIQAHIGAYDAIKEYSEKSVGVIYAFAWHDPLAEEYKDEVEEIRKKDYEFVTILHSKGKLDWIGVNYRLVYGAKDGHLVPLPGYGFMSERGGFAKSGRPASDFGWEMYPEGLENLLKYLNNAYELPMIITAVYNAMKEGADVRGYLHWSLTDNYEWAQGFRMRFGLVYVDFETKKRYLRPSA |
| 2 | 6gmhQ | 0.06 | 0.06 | 2.64 | 0.50 | EigenThreader | | GGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGVEAMQAESCYQLARSFHVQEDYDQAFQYYYQATQFASSSFVLPFFGLGQMYIYRGDKENASQCFEKVLKAYPNNYETMKILGSLYAASEDQEKRDIAKGHLKKVTEQYPDDVE |
| 3 | 6y9bC | 0.14 | 0.12 | 4.15 | 0.49 | FFAS-3D | | LF--PQWHLPIKIAAIIASLTFLYTPLATSHQQYFYKIPILVINKVLPMVSI--TLLALVYLPGVIAAIVQLHNGTKK--FPHWLDKWMLTRKQFGLLSYPMRRSYRYKLLNWAYQQVQQNKEDAWIEHDVWRMEIYVSLGIVGLAILALLAVTSIPSVS--------------------DSLTWREFHYIQSKLGIHNKWIDIKQFVWYTPP- |
| 4 | 5jcss | 0.08 | 0.07 | 2.95 | 1.09 | SPARKS-K | | MIGMRIWNVEEPSEEDLTHILAQKFPILTNLIPKLID--SYKNVKSIY-----MNTKFISLNKG-------AHTRVVSVRDLIKLCERLDILFKNNGINKPDQLIQSSVYDSIFSEAADAGAIGEFKALEPIIQAIGESLDIASSRISLFLTQHVPTLENLDDSIKEKLNIQKKSMNSTLFAFTNHSLRLMEQISVCIQMTEPVLLVGETGTGK |
| 5 | 2ynaA | 0.21 | 0.04 | 1.34 | 0.26 | CNFpred | | ----NEWALSN----QFTEFVGTQSIDMLAHRTGVSVEQMLAAIQSLHAGF------------------------------------------------------------------------------------------------------------------------------------------------------------------- |
| 6 | 7bgyB | 0.06 | 0.05 | 2.00 | 0.83 | DEthreader | | VWIGSLLTTCISIAMASGA---NALFAAISGWLWITVLFANFAEALAEGRSK--AQASLKGTFLDR-MIAMVEGAQR--RKTPNEIALTILLIALTIVFLATALVCLIP-TTIGGLLSAI-GVAGM-------------------------AVEAADVDVVAMTGLAKEA----MVDLSKLEVVHIGKQMLMGPLTA----------------- |
| 7 | 3oa5A | 0.09 | 0.09 | 3.52 | 0.76 | MapAlign | | IAVRLGVATDPDDAIANHKGKTIPVDPDGAVLASEKAKGLLGGFRLLHEASMSGLFSEIAKDEILRTNFVEGIKDFFQRFPMFSHLDIDWEYPGSIGAGNPNSPDDGANFAILIQQITISIASSADPAKIDAALKGTYTDANQTLGSFEYSVLEWTDIICHYMDFEKGEGRNGYKLKVFISLDTPRSVRDKGRYVKDKGLGGLFIWSGDQDNGI |
| 8 | 6fmlG | 0.11 | 0.11 | 3.90 | 0.87 | MUSTER | | GYSTRSLVEYRLPRLIWCDGGRLDKPGPGNLVAGFRSKYLNHPENIRSSLEGIENFTWLRFVDTSLQEAYRASHTDVFASKQNRLGHMQIVYDEPEDKKWTPVLFQICERENPKAVAEITTEGVLRDLMNIARVKYRELGLCRLEKAARPRASAPPIEVVCDSRSAVIERENIMFHPAMRKALFGPT-PSEIKEASFGPRPVTLYPPRALLPAP |
| 9 | 2pffB | 0.18 | 0.16 | 5.35 | 0.54 | HHsearch | | GLNILEWLENPSN-TPDKDYLLSIPIS-CPLIGVIQLAHYVVTAKLLGFTPGEL-RSYLKETDSWESFVSVR----------KAI-TVLFFIGVRCPNTSLPPSILEDSLENEGVPSP-MLSISNLTQEQQYVNKTNSISLVNGAKNLVGLNLTLRKAKAPSGLDQSRIPFSEVASPFHSHLLVPADLINKDLVKNNVSFNAKDIQIPVYDTFD |
| 10 | 6l7dA | 0.10 | 0.10 | 3.65 | 0.34 | CEthreader | | EAVELRDGGDRYGGKGVQKAVQAVLDEIGPAVIGLNADDQRLVDQALVDLDGTPDKSRLGGNAILGVSLAVAKAAADSANILNGGAHADTAVDIQEFMVAPIGAPSFVEALRWGAEVYHALKSVLKKEGLSTGLGDEGGFAPDVAGTTAALDLISRAIESALDAAATEFFTDGTGYVFEGTTRTADQMTEFYAGLLGAYPLVSIEDPLSEDDWD |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|