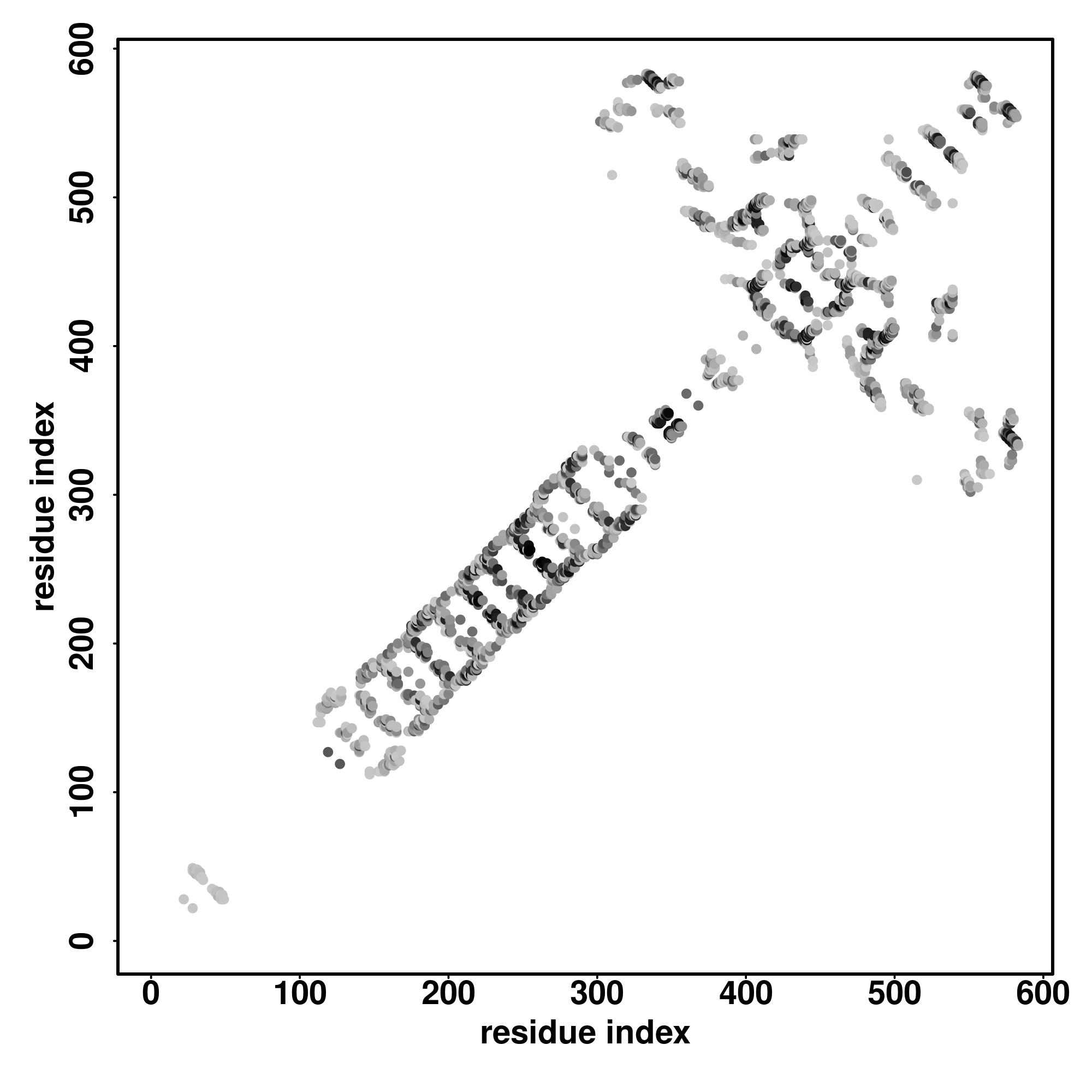
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200
| | | | | | | | | | |
|---|
| SS
Seq | CCSSHHHHHHHHHHHCCCCCCCCCCCCCCCHHHHCSSSSCCCCSSSSSCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCHHHHHHHHHHHHHCCCCCCCCCCCCCCCCCCCCCCCCCHHHHHHHHHHHHHCCCHHHHHHHHHHHHHHHCCCCHHHHHHHHHHHHHHCCCCCHHHHHHHHHHHHHCCCHHHHHHHHHHHHHHCC
MTFYLFGIRSFPKLWKSPYLGLGPGHSYVSLFLADRCGIRNQQRLFSLKTMSPQNTKATNLIAKARYLRKDEGSNKQVYSVPHFFLAGAAKERSQMNSQTEDHALAPVRNTIQLPTQPLNSEEWDKLKEDLKENTGKTSFESWIISQMAGCHSSIDVAKSLLAWVAAKNNGIVSYDLLVKYLYLCVFHMQTSEVIDVFEIMKARYK |
|---|
| 1 | 5iwwD | 0.09 | 0.09 | 3.28 | 0.56 | CEthreader | | MYSKCGLLEEARKVFDEMPEKDVVTYNTLIDGLCKAGKLDEALKLFEEMVEKGIKPDEFTFSSVLKACARLGALELGKQIHGYVIKSGF------------ESNVVVYNALIDMYSKCGLLEEARKVFDEMPEKD--VVTYNTLIDGLCKAGK-LDEALKLFEEMVEKGI-KPDEFTFSSVLKACARLGALELGKQIHGYVIKSGF |
| 2 | 5zyqA | 0.05 | 0.04 | 2.12 | 0.65 | EigenThreader | | DEVIELDFDQLEVISILKQEHTQLHIWIALALEYYKQGKTEEFVKLLEAAGNLDYRDLDTLAAYYVQQARKEKNDLITQATLLYTMADKGRACFCLLEGDKFVLNQSPNNIPALLGKACISFNK----------KDYRGALAYYKKALRTNPCPAEVRLGMGHCFVKL---NKLEKARLAFSRALELNSKCVGALVGLAVLEKHDL |
| 3 | 2g2bA | 0.12 | 0.07 | 2.41 | 0.53 | FFAS-3D | | ---------------------------------------------------------------------ESQTRDLQGGKAFGLLKAQQEERLDEINKQ--------FLDDPKYSSDEDLPSKLEGFKEKYMEFDLNDIMSLKRMLEKLGVPKTHLELKKLIGEVSSGSGETFSYP---DFLRMMLGKRS--AILKMILMY----- |
| 4 | 4bj1A | 0.10 | 0.09 | 3.40 | 0.73 | SPARKS-K | | QKFKSMALQELGTNYLSIYVPLRSMKNCIVFFDKVEYLMKDHIVYFKFEQLDKIEPSKLTEFINVLSVLEKSNN----IAFKVLIYSNSSLLSTSLKKKLN-----TKYTVFEMPI--LTAQEQEYLKKMFDSGSKLQSYNSLVTCQLNNKES------NLAIFFEFLKVFPHPFTYLFNAYTEIIVQSRT--FDELLDKIRNRLT |
| 5 | 5x9aA | 0.10 | 0.05 | 1.80 | 0.62 | CNFpred | | ----------------------------------------------------------------------------------------LAEELYKTSCQ-----------------KHFTKTEVESLIICYKNLLEGLKMDRNLFRDILHQKF-DLLMDRVFRAFDKDSDSYISLTEWVEGLSVFLRGTLDEKMEYTFTVFDLNGD |
| 6 | 7dxjA | 0.03 | 0.03 | 1.63 | 1.00 | DEthreader | | ----PPEKEKDVKFVVALGDCCALQLP-GL----------------WSVVSHACSLIHCVHFILEAVVQPG---------YIAAEMVAEVATYRINTLGASLSYKLPHGVVLIPVISDYLLSNLKGIAC--HSQQHVLVMCATAFYLIENYPLGPEFSASIIQMCGVMSGSTIYHCALRGLERLLSEQLSRLDAESLVKLSVDRVN |
| 7 | 4ui9X | 0.08 | 0.08 | 3.08 | 0.87 | MapAlign | | KEVLRQCPLALDAILGLLSLSVKGAEVASMTMNVIQTVPNLDWLSVWIKAYAFVHTGDNSRAISTICSLEKKSLLRDNVDLLGSLADLYSVLKFEQAQMLDPYLIKGMDVYGYLLAREGRLEDVENLGCRLFNISDHAEPWVVSGCHSFYSKRYS-RALYLGAKAIQLN--SNSVQALLLKGAALRNMGRVQEAIIHFREAIRLA- |
| 8 | 2yr4A | 0.08 | 0.07 | 2.91 | 0.48 | MUSTER | | MRFPEIA-GLTWHYAS--AFGDAAPIKVFPNPGKVPTEFVFGNRVDRYVGSDPEDPDSPTLKVLGVVAGGLVGNPQGENVAMYPIANVDPAKIAAILNAATPPADALER---------IQTKYWPEFIAQYDGLTLGAAVREIVTVAFEKGTLPPESISYYVELFGRFGFGTGGFKPLLVEMMRLILWDYSNENVEFIRNLFLKAQ |
| 9 | 5dizA | 0.12 | 0.04 | 1.37 | 0.90 | HHsearch | | ---------------------------------------------------------------------------------------------------------------------------------------NPETNLLFNLNSCSKSK-DLSAALALYDAAI-TSSVRLHFQTLL---YLCSASITDIRGFEIFDRMVSSGI |
| 10 | 6vbu4 | 0.08 | 0.08 | 3.10 | 0.52 | CEthreader | | FLLKGDLDKAIEIYKKAVEFSPENTELLTTLGLLYLQLGIYQKAFEHLGNTLTYDPTNYKAILAAGSMMQTHGDFDVALTKYKVVAPPLWNNIGMCFFGKKKYVAAISCLKRANYLAPLDWKILYNLGLVHLTMQQYASAFHFLSAAINFQPKMGELYMLLAVALTNLEDSENAKRAYEEAVRLDKCNPLVNLNYAVLLYNQGEKR |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|