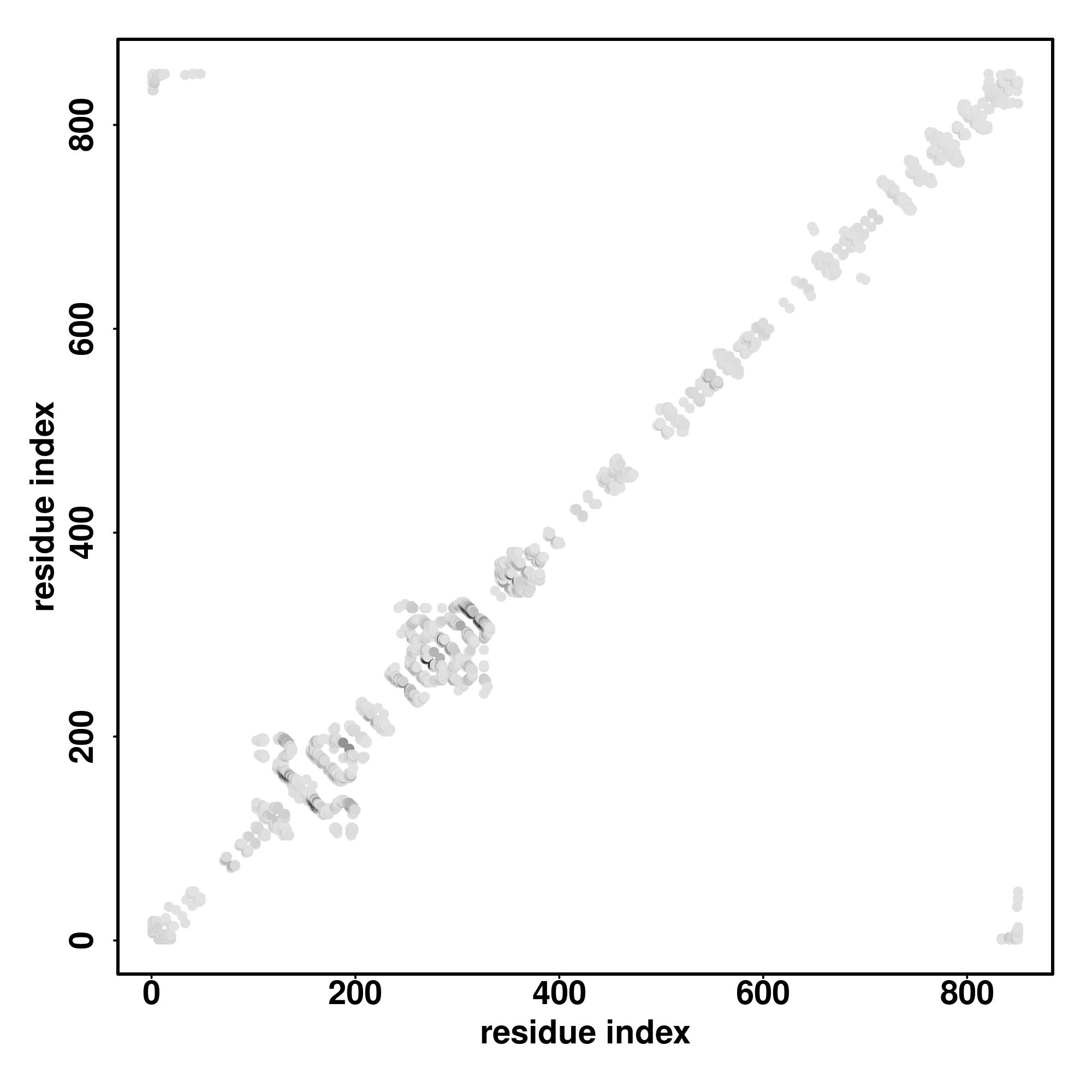
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180
| | | | | | | | | |
|---|
| SS
Seq | CSSSSSSSSSSSSSSSSSCHHHHHHHHHHHHHHHCCCCCCCCCCCCCCCCCCCHHHCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCC
ICVALLVVGIVCVVAYCKTKKQRKQMHNHLRQNMCPAHQNRSLANGPSHPRLDPEEIQMADYISKNVPATDHVIRRETETTFSGSHSCSPSHHCSTATPTSSHRHESHTWSLERSESLTSDSQSGIMLSSVGTSKCNSPACVEARARRAAAYNLEERRRATAPPYHDSVDSLRDSPHSERYVS |
|---|
| 1 | 1vt4I | 0.05 | 0.05 | 2.33 | 0.59 | CEthreader | | SILNTLQQLKFYKPYICDNDPKYERLVNAILDFLPKIEENLICSKYTDLLRIALMAEDEAIFEEAHKQVQRGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG |
| 2 | 5tj5A | 0.04 | 0.04 | 2.18 | 0.53 | EigenThreader | | GGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGREINAGLPTIVTFPFMFAIMFGHGFLMTLAALSLVLNTGRYIILLMGVFSMYTGFLYNKMKLSILMGFIHMTYSYFFSLANHLDIIGNFIPGLHTASYLRLWALSLAHAQLSS |
| 3 | 6em3x | 0.07 | 0.04 | 1.57 | 0.35 | FFAS-3D | | ------------------NKLKRQEIFADIKHEKNKER--------------HTMRRKRAKEERENPELREQRLKENVTQTIENTRVYEDDLMRYFNSNSNEPP---------KIFLTTNVNAKKSAYEFANILIEILP-------------------------------------------- |
| 4 | 5lj3W | 0.05 | 0.04 | 1.85 | 1.26 | SPARKS-K | | -----------------------MKFTPSIVIDAPQYYVDHFNGKYNVDKCVILRDLQL----ETDSESMPSSLKHLTHIL----DLTNNDLIMIPDLSRRDDIHTLLLGRNNIVEVDGRLLPMNVQNLTLSNNSIRRFEDLQRLRTLKNLTNYREHVLRLVPHLETLDFQNVTAEERKSA-- |
| 5 | 2po3A | 0.14 | 0.03 | 0.94 | 0.42 | CNFpred | | MSEAAAAMGLTSLDAFPEVIDRNRRNHAAYREHLADL-------------------------------------------------------------------------------------------------------------------------------------------------- |
| 6 | 5y9dA | 0.07 | 0.05 | 2.26 | 0.83 | DEthreader | | ---VHG-K-D-YGT-RNDQLTYGALIRGRVSMIAD-FHKRLTIALRYAVRRQFETKIIDPQRLLPLLAYCMKMGADEAKTIETTDRILALNQDLEKAVDTKELFAAAGMKAFTWCA-IDRQGYSG-F---------C--DNNVLCLSMG--NPDQAELSQQFQ------EE--V-C------- |
| 7 | 1vt4I | 0.04 | 0.04 | 2.02 | 0.97 | MapAlign | | ---TLQQLKFYKPYICDNDPKYERLVNAILDFGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG |
| 8 | 4k0mC | 0.11 | 0.10 | 3.71 | 0.72 | MUSTER | | -----------------KTIDEAETVEVHAKLGIDPRRSDQNVRGTVSLPHAKGEKIKEAEEAGADYVGGEEIIQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA |
| 9 | 2pffB | 0.20 | 0.17 | 5.42 | 0.75 | HHsearch | | KAIVLFFIGVRCYEAYPNTSLPPSILEDSLENNEGVPSP--MLS----ISNLTQEQVQ--DYVNKAKAPSDQIPFSERKLKFSNRFLP--------------------VASPFHSHLLVSDLINKLVKNNVSFNAKDIQICIRLPTQFKA-THILDFGPGGLTHRKDIVAGTLDINPDDDYGF |
| 10 | 3b98A | 0.08 | 0.08 | 3.05 | 0.39 | CEthreader | | TQIYEEFRRFDKLLPKLARTTVNKEEKQIASAAREKLWKWLTPSGLDRKPREQSWLGSYVKQLQDEGIDAEMQRRAMLLQLWVTQGNAGPA---AFWVMGYLLTHPEALRAVREEIQNTPVFDSVLWETLRLTAAALITRDVTQDKKICLSNGQEYHLRRGDRLCVFPFISPQMDPQIHQQPE |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|