| |||||||||||
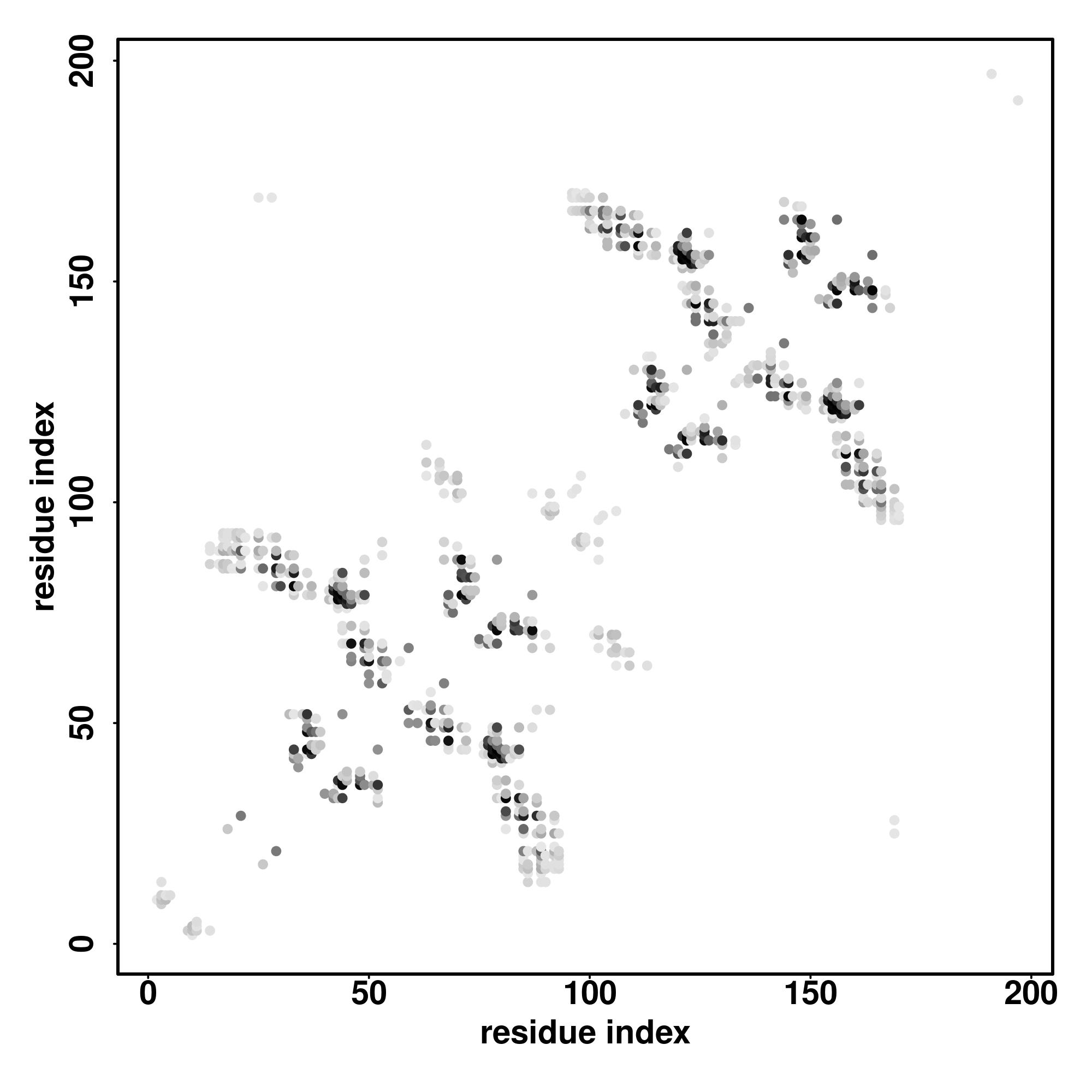
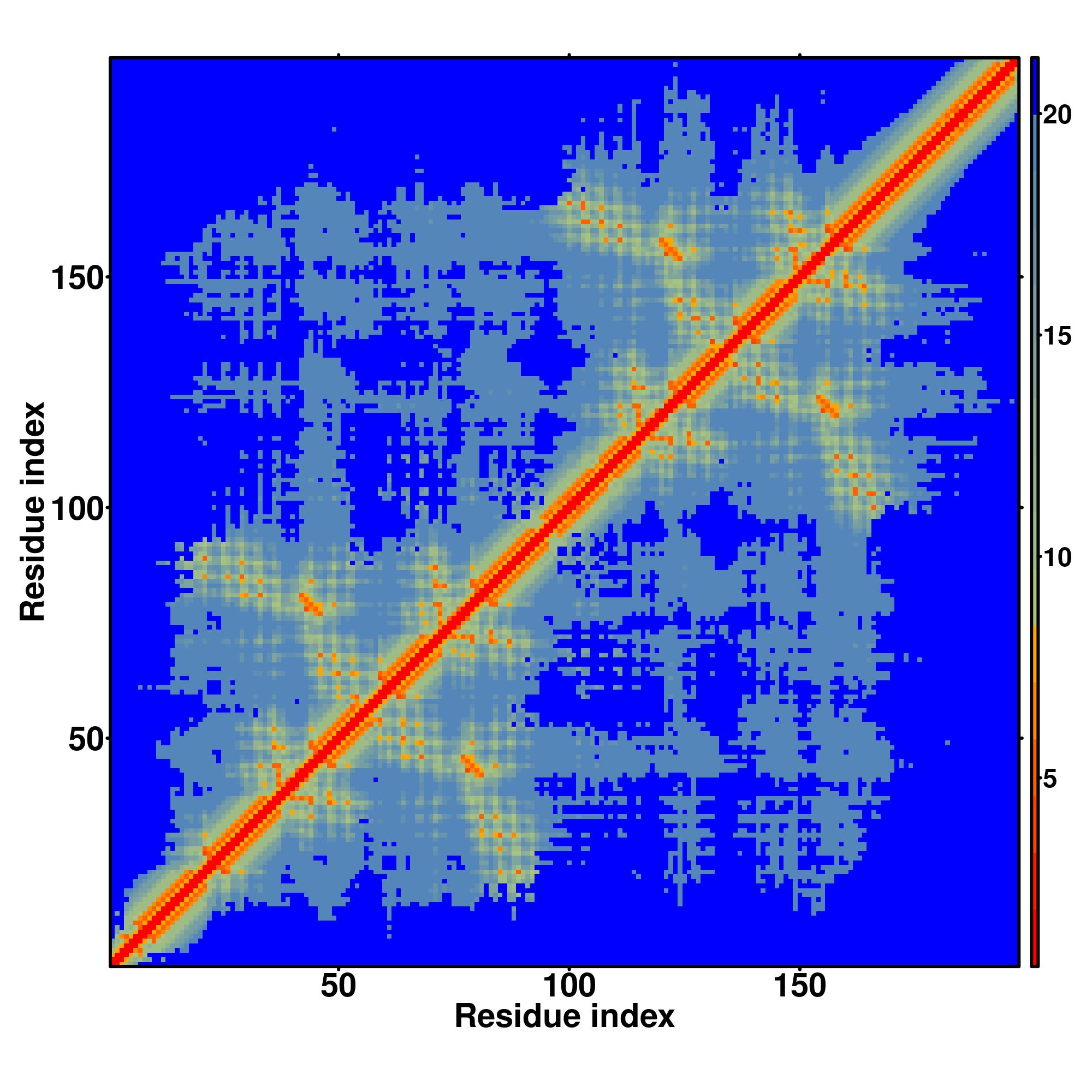

| D-I-TASSER simulation is guided by the consensus contact map (left figure), distance map (middle figure) and Hydrogen bond network (right figure) derived based on confidence scores of AttentionPotential. In the contact, distance map and hydrogen bond networks, the axes mark the residue index along the sequence. For the contact map, each dot represents a residue pair with predicted contact, while for the distance map and hydrogen bond network, a color scale represents a distance of 1-20+ angstroms or a angle of 0-180 degree. |


