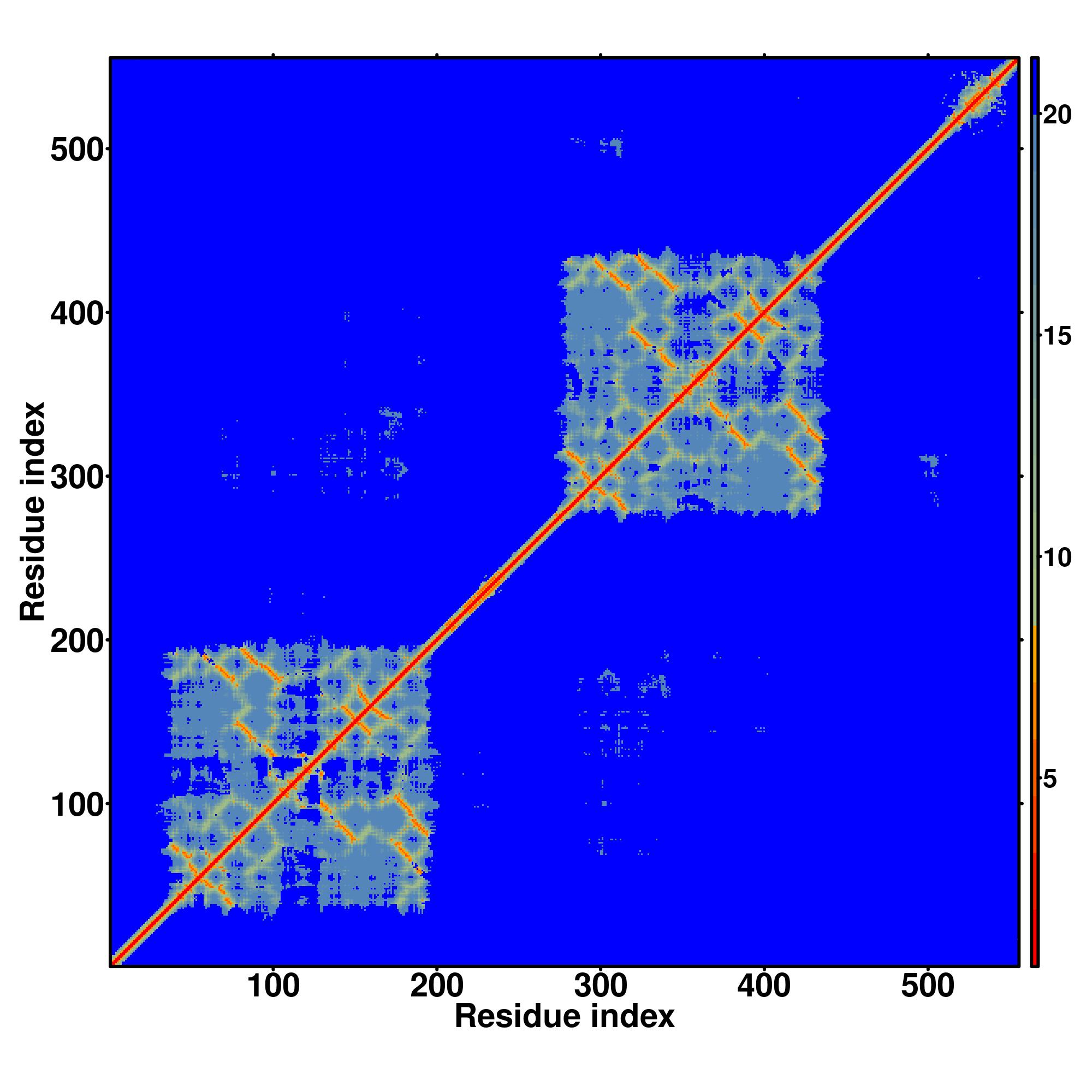
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180
| | | | | | | | | |
|---|
| SS
Seq | CCCCCCCCCCCCCCCCCCCCCCCCCCCCCSSSCCCCSSSSSCCCCCCCSSSSSCCCCCCCCSSSSSSSSCCCCCSSSSSSSSSSCCCCCCCCCCCCCCHHHHHCCCCCSSSSCCCCCCCCCCSSSSSSSCCCSSSSSSCCCCCSSSSSCCCCCCSSSSSSSSCCSSSSSSSSCCCCCCCCCCCCCCCCCCCCC
CGPRERPRPASSPALLEADLRFHATRGPDVSLSADRKVACAPRPDGGRTLVFSERPLRPGESLFVEVGRPGLAAPGALAFGITSCDPGVLRPNELPADPDALLDRKEYWVVARAGPVPSGGDALSFTLRPGGDVLLGINGRPRGRLLCVDTTQALWAFFAVRGGVAGQLRLLGTLQSSPATTTPSGSLSGSQD |
|---|
| 1 | 2yueA | 0.25 | 0.21 | 6.58 | 1.17 | DEthreader | | ----------------SSGLQFHSVHGDNIRISRDGTLARRFE-SFCRAITFSARPVRINERICVKFAEISNNWNGGIRFGFTSNDPVTLEGTLPKYACPDLTNRPGFWAKALHEQYCEKDNILYYYVNGAGDVIYGINNEEKGVITGIDTRSLLWTVIDIYGNC-TGIEFLDSRIYMYQ------------- |
| 2 | 2yueA | 0.27 | 0.23 | 7.02 | 2.73 | SPARKS-K | | -----------GSSGSSGPLQFHSVHGDNIRISRDGTLARRFE-SFCRAITFSARPVRINERICVKFAEISNNWNGGIRFGFTSNDPVTLEGT-LPKYAPDLTNRPGFWAKALHEQYCEKDNILYYYVNGAGDVIYGINNEEKGVILTIDTRSLLWTVIDIYGNC-TGIEFLDSRIYMYQ------------- |
| 3 | 5ji7A | 0.13 | 0.10 | 3.41 | 1.18 | MapAlign | | ------------------LPSWSPDKFSYIGLSQNNLRVHYKGHPKDAASVRATHPICGIYYFEVKIVSKGR--DGYMGIGLSA--QGVN----M-NRL--PGWDKHSYGYHGDGPTFTTGDVIGCCVNLINTCFYTKNGHSLGIAFTLPP--NLYPTVGLQ-TPGEVVDANFGFVF---------------- |
| 4 | 2yueA | 0.25 | 0.22 | 6.74 | 1.02 | CEthreader | | -----------GSSGSSGPLQFHSVHGDNIRISRDGTLARRFE-SFCRAITFSARPVRINERICVKFAEISNNWNGGIRFGFTSNDPVTLEGTLPKYACPDLTNRPGFWAKALHEQYCEKDNILYYYVNGAGDVIYGINNEEKGVILTIDTRSLLWTVIDIYG-NCTGIEFLDSRIYMYQ------------- |
| 5 | 2yueA | 0.27 | 0.23 | 7.02 | 2.20 | MUSTER | | -----------GSSGSSGPLQFHSVHGDNIRISRDGTLARRFE-SFCRAITFSARPVRINERICVKFAEISNNWNGGIRFGFTSNDPVTLEG-TLPKYAPDLTNRPGFWAKALHEQYCEKDNILYYYVNGAGDVIYGINNEEKGVILTIDTRSLLWTVIDIYGNCT-GIEFLDSRIYMYQ------------- |
| 6 | 2yueA | 0.27 | 0.23 | 7.02 | 5.85 | HHsearch | | -----------GSSGSSGPLQFHSVHGDNIRISRDGTLARRFE-SFCRAITFSARPVRINERICVKFAEISNNWNGGIRFGFTSNDPVTLEG-TLPKYAPDLTNRPGFWAKALHEQYCEKDNILYYYVNGAGDVIYGINNEEKGVILTIDTRSLLWTVIDIYGNC-TGIEFLDSRIYMYQ------------- |
| 7 | 2yueA | 0.27 | 0.22 | 6.68 | 2.03 | FFAS-3D | | -----------------GPLQFHSVHGDNIRISRDGTLARRFE-SFCRAITFSARPVRINERICVKFAEISNNWNGGIRFGFTSNDPVTLEGTLPKYACPDLTNRPGFWAKALHEQYCEKDNILYYYVNGAGDVIYGINNEEKGVILTIDTRSLLWTVIDIYGNC-TGIEFLDS------------------- |
| 8 | 2yueA | 0.25 | 0.21 | 6.60 | 1.33 | EigenThreader | | -----------GSSGSSGPLQFHSVHGDNIRISRDGTLARRFE-SFCRAITFSARPVRINERICVKFAEISNNWNGGIRFGFTSNDPVTLEGTLPKYACPDLTNRPGFWAKALHEQYCEKDNILYYYVNGAGDVIYGINNEKGVILTGIDTRSLLWTVIDIY-GNCTGIEFLDSRIYMYQ------------- |
| 9 | 4kg0A | 0.29 | 0.23 | 6.94 | 2.36 | CNFpred | | -------------------LQFHSVHGDNIRISRDGTLARRFES-FCRAITFSARPVRINERICVKFAEISNNWNGGIRFGFTSNDPVTLE-GTLPKYAPDLTNRPGFWAKALHEQYCEKDNILYYYVNGAGDVIYGINNEEKGVILTIDTRSLLWTVIDIYGNC-TGIEFLDS------------------- |
| 10 | 2fnjA | 0.07 | 0.06 | 2.56 | 1.17 | DEthreader | | FV--DILDMPPASRDLQLKHSWNEDRSLNIFVKEDKLTFHRHPVAQSTDCIRGKVGLTGLHIWEIYWP-TRQRG-THAVVGVC-TA--DA---PLHSQSLVGSTE-QSWGWDLRNFLVP--DKFLVALDDEGTLSFIVDQQYLGIAFRGLRGKKLYPIVSAV-WGHCEITMRYIGGLDLPDLC-R-------- |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|