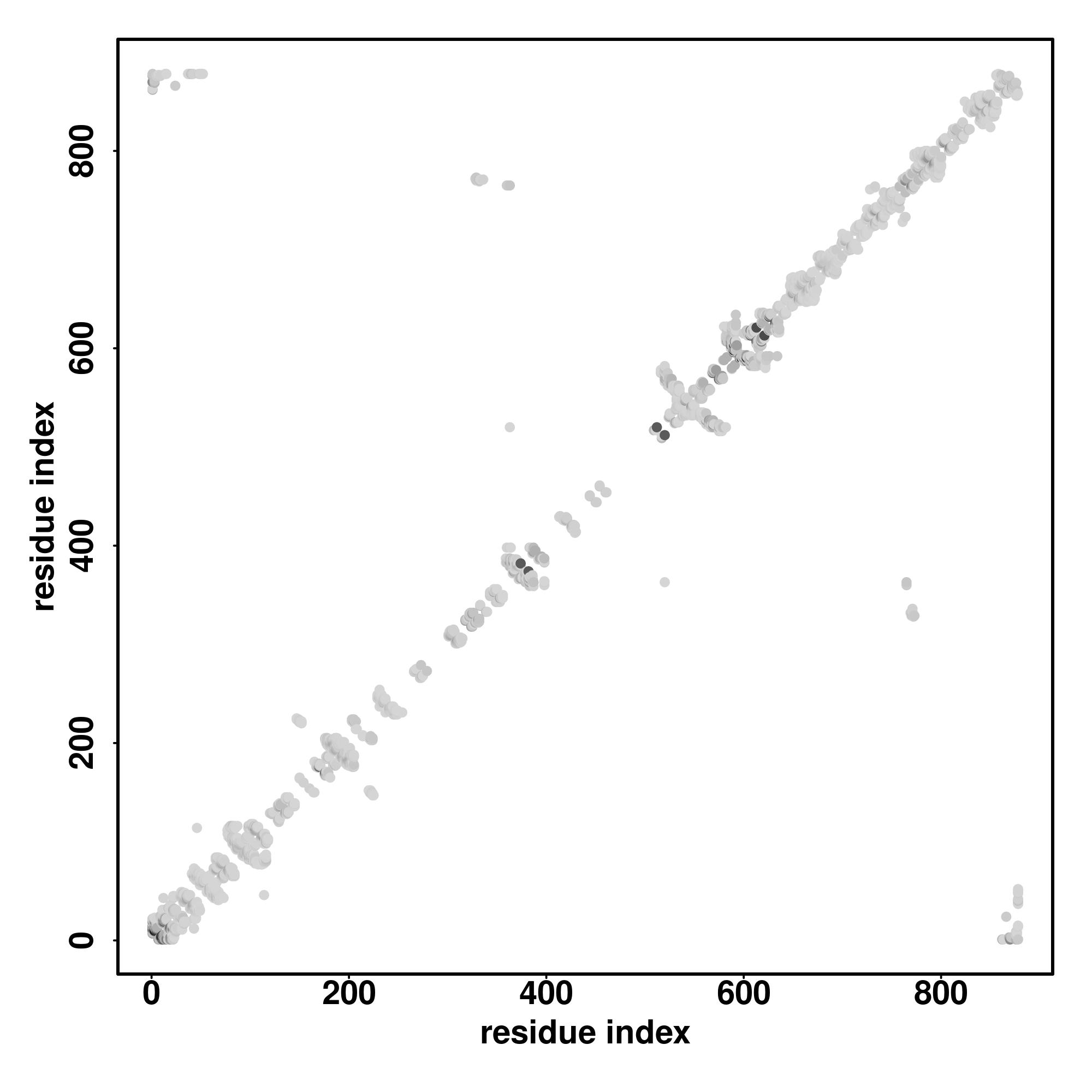
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240 260 280 300 320 340 360 380
| | | | | | | | | | | | | | | | | | | |
|---|
| SS
Seq | CCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCHHHHHHHHHHHHHHHHHHCCCCCCCCCCSSSSSSSCCCCCCCCCCCCHHHCCCHHHHHHHCCCCCCHHHHHHHHHHHHHHHHHHHHCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCHHHHHHHHHHHHHCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCHHHHHHHHHHCCCHHHCCCCCCCCCHHHHHHHCCCCCCCCHHHHHHHHCCCCCCCHHHHHHHHHHCCCCCCCCCCCCCCCCCCC
QVVGGTQACEGGWSQGLPLPPPPPPAAQLPPIVSQGNAGPWPQGAHGESSLASSQAKAPPDDSCNPRSVYENFRLWQHYKPLARRHLPQSPDTEALSCFLIPVLRSLARRKPTMTLEEGLWRAMREWQHTSNFDRMIFYEMAEKFLEFEAEEEMQIQKSQWMKGPQCLPPPATPRLEPRGPPAPEVVKQPVYLPSKAGPKAPTACLPPPRPQRPVTKARRPPPRPHRRAETKARLPPPRPQRPAETKVPEEIPPEVVQEYVDIMEELLGPSLGATGEPEKQREEGKVKQPQEEDWTPPDPGLLSYIDKLCSQKDFVTKVEAVIHPQFLEELLSPDPQMDFLALSQDLEQEEGLTLAQLVEKRLPPLKEKQHARAAPSRGTARL |
|---|
| 1 | 5yfpC | 0.07 | 0.07 | 2.96 | 0.74 | CEthreader | | EYPTNKGLYQEIMSGTISTRTAPRGYKHFLINGINNSISEMFGEMREKYIFNELIIVKEHIANCCPPHWNIFEVYFDQYYKELHSLITDLVESEPETIIILDILAFDKTFQDTLKQDKSVIGDKEKETLFKDYLNLIVVKMTEWIGNLEKAEFDVFLERSTPPHSDSDGLLFLDGTKTCFQMFTQQVEVAAGTNQAKILVGVVERFSDLLTKRQKNWISKISEEIKKQINYNHKYDIDPESITPEDECPGGLVEYLIAVSNDQMKAAD--YAVAISSKYGKLVSKVYEKQITNHLEGTLDGFAEVAQCSS--LGLITLMFDDLRKPYQEIFSKTWYMGSQAQQIADTLDEYLLDIKPQMNSVLFVNFIDNVIGETIIKFLTAL |
| 2 | 4uzyA | 0.07 | 0.07 | 2.90 | 0.80 | EigenThreader | | PSNEDYKLYYAQSLYKGGMYPEASKAVVKVEGHQKAVTTLLVACSYEQDDLTGCRRQLDKCAPEDPDTMVNTGCIMFKELYNIALCYYKTKQFGPALKHLAEIIEKAVREHPELSLIEAFNLKAAIEYTMKNVEAAKEALTDMSALINTGGFKKLNFLLQSPPFPPETFANLLLLYCKPSHGFYDLAADVLAENPQYAGKLLSPDLYDYLQAAIGRYKSPEEAFRRFDELATRHVEQLRRLTKQIQDARIARDNDAIKRAINEYDEALEAYIPGLMAMASIYWDMELIFRQSAEFCSEHEVWKLNVAHTFFMQDNHYKEAIRYYEPVVKKNADNLLGVTAIVLANLCVSYIMTHLCIINLVIGTLYCAKGNYEFGVSRIIKSL |
| 3 | 3dl8G | 0.12 | 0.10 | 3.63 | 0.71 | FFAS-3D | | -PIPGINFALYDIFSGGNLGRLTVFALGVMPYISAS---------IMMQLLTVAIPSLQRLAKEEGDYGRYKINEYTKYLTLFVAT------VQSLGIAFWFILITVLTLVAGTMFLVWIADRITEKGIGNGASLIIFAGIVANFLTLLLIIALIIAIIVGIVYVQEAERRIPIQYPGRQVGRQLYAGRKTYLPIKINPAGVIPII--------------FAQALLLIPSTLLNFVQNPFIKVIADMFQPGAIFYNFLYVTFIVFFTY--FYTAVLINPVELAENLHKAGAFIPGVRPGQDTVKYLERIINRLIFFGALFLSVIPILISVWFNIPFYFGGTTAL--IVVGVALDTFRQIETYLIQKKYKSYVR---------- |
| 4 | 4btgA | 0.11 | 0.10 | 3.74 | 0.81 | SPARKS-K | | SARGLTQAFAIGELKNLSVGALQLPTRTFSASMTSELLWEVGKGNIDPVQYAQAGGALSADAVVPPTAILEQLRTTDFVCHVLSPRTATYPNVDCVRASDLRMLTALSSLQATFKAKGALALISQHLANAATVVSSVLTILGRLWLRSNLALFIAYQDMVKQRGR---AEVIFSDEELSSTIIPWFIEAMSEVSPFKLRPINSYIGQTSAIDHMGQPSHVVVYEDWQFAKEITAFTPVKLANNSNQRFLDVEP-----GISDRMSATLAP-IGNTFAVSAFVKNRTAEAVSQRGTVNSNG------------------AEMTLFPSVVERDYALD--RDPMVAIAALRTLKRSMFNYYAAVMHYAVAHNPEVVVSEHQGVAAE |
| 5 | 6g90O | 0.13 | 0.07 | 2.53 | 0.76 | CNFpred | | -------------------------------------------------------------------AVVAKALGVNQLLPFINAACHS--KSWKARHTGIKIVQQIGILLGVLNHLTGLMSCIKDCLMDHVPVRIVTAHTLSTLAENSGIEVFNVVLEPLWKGI-----------------------------------------------------------------RSHRGKVLSSFLKAVGSMIPLMDPEYAGYYTEAMRIIRREF----DSPDDEM----------------KKTILLVLQKCSAVESITPFLREEIAPEFFQKFWALDRPLNKVVTYTTVTLAKKLGCSYTIDKLLTPLRD--------------- |
| 6 | 7b52A | 0.06 | 0.03 | 1.41 | 0.67 | DEthreader | | -------------------------------------DNY----------RSKLDGNDVTFFNLFEQNLFLFFSCWEEYIQKYFNSKNIGSDTFEFLIKKCGNIFSEKLKNAEKK--K---------TE-----------------------------------------------------------------------------------------QGACKRKCEKYKKYISCISANYKKLEGRSLEG-V-YVPPRRQQLDDHYTKYIDSKLNEIFGS----SNT-N--D-I--TKRRT---IAKPQFIRLEEWTNEFCEKYTKYFEDMKSKCIECKCANYTNWLNPKRIEWN-M--ICKNIGALEDKKYSHKMKCT-Y-- |
| 7 | 1vt4I | 0.05 | 0.05 | 2.33 | 0.97 | MapAlign | | RRLLKSKPYENCLLVLLNVQNAKAWNAFNLSCKILLTTRFKQVTDFLSAATTTHISLDHHSMTLTPDEVKSLLPRRLSIIAESIRDGLATWDNLTTIIESSLEYRKMFDLSLIWFDSDVMVVVNKLVKLENEYALHRSIVDHYNIPKTYFYSHIGHHLKNIEHPERMTLFRMVFLDFRFLEQKIRHDSTAWNASGSILNTLQQLKFYKPYICDNDPKYERLVNAILDFLPKIEENLICSKYTDLLRIALMAEDEAIFEEAHKQVQGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG---GGGGGGGGGGGGGGGGGGGG |
| 8 | 4btgA3 | 0.13 | 0.12 | 4.07 | 0.68 | MUSTER | | SARGLTQAFAIGELKNQLSVGALQLPLQFTRTFSASMTSELLWEAY---VYRVGRTATYPFDA--------NAVVSSVLTILGRLWSPSTP----------------KELDPSARLRNGIDQLRSNLALFIAYQDMVKQRAEVIFSDEELSSTI-IEAMSEVSPFKLRPINETTSYIGQTSAIDHMGQPSHVVVYEDWQFAKEITAFTPVKLANNSNQRFLDVEPGISDRMSATLAPIGNTRGTVNSNGAEMT--GFPSVVERDYALDRDPMVAIAALRTGIVDESLEARA-----------SNDLKRSMFNYYAAVMHYAVAHPEVVVSEHQGVAEQGSLYLVWNVRTELRIPVGYRTPEPLEAIAYNKPIQPSEVLQAKVL |
| 9 | 5vweA | 0.15 | 0.03 | 0.88 | 0.59 | HHsearch | | --------------------------------------------------------------------------------------------MSAYMLWLNASREKIKSDHPGISITDLSKKAGEIWKGMSKEKKEEWDRKAEDARRD-YEKA----MKEYEGG--------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------- |
| 10 | 1vt4I3 | 0.05 | 0.05 | 2.41 | 0.70 | CEthreader | | GGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|