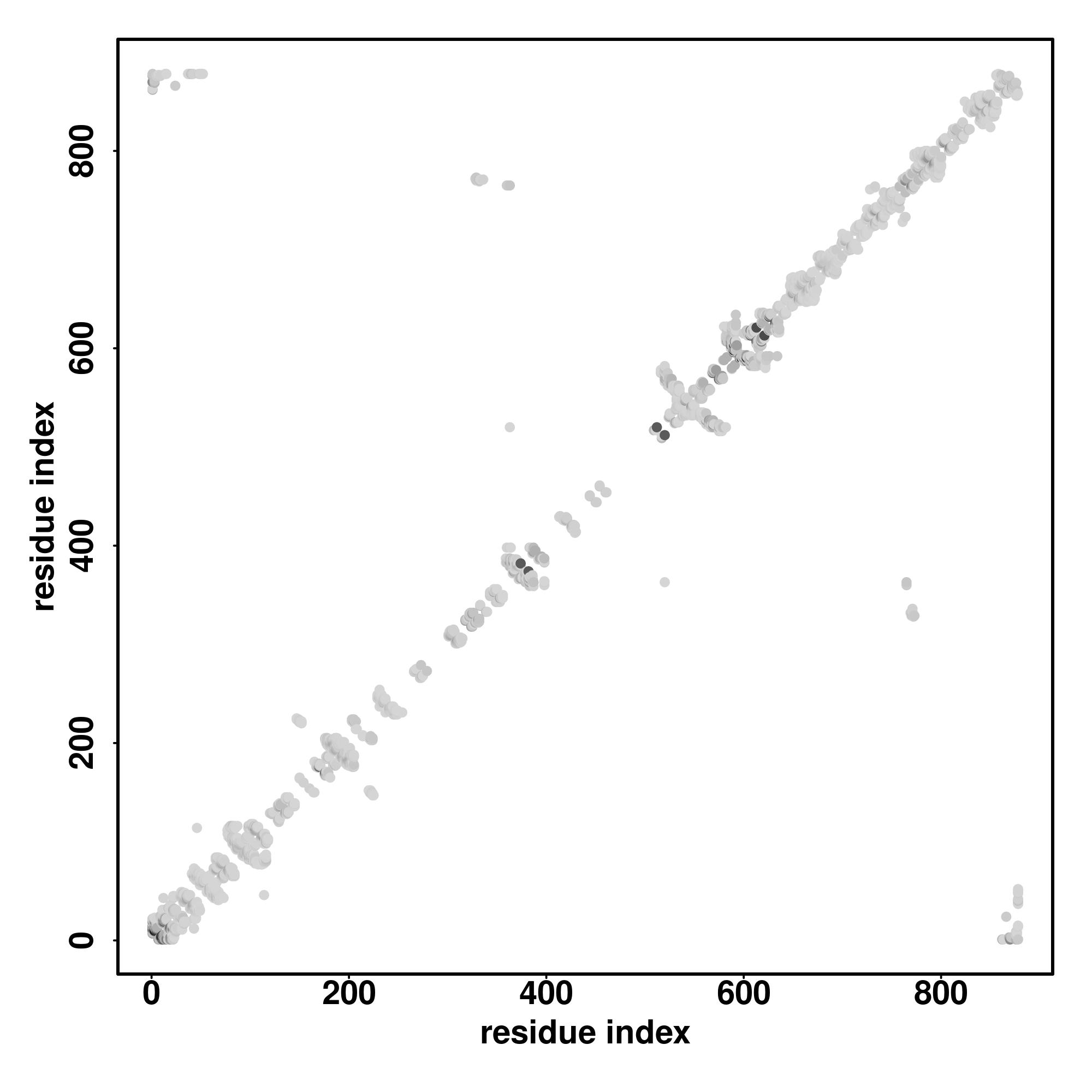
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240 260
| | | | | | | | | | | | | |
|---|
| SS
Seq | CCCCCCCCCCSSSCCCCHHHHHHHCCCCCSSSCCCCCCCCCCSHHHHHHHHCCCCCCCCCCCCCCCSSSSSCCCCCSSSSSSSSSCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCSCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCSCCCCCCCCCCCCCCCCCCCCCCCCCSSSSSCCCCCCCCCCCCCSSSSSCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCC
MEVKGPSGRSFCCESEGQFKSCLKRHTPSLLLPSSWKGNSGSCLMAEALHRTSPTPNSCPLPLPLCRMSGVLCSRNLFTFKFSLFQLDSGASGEPGHSLGLTLGFSYCGNCQTAVVSAQPEGMASNGAYPVLGPGVTANPGTSLSVFTALPFTTPAPGPAHGPLLVTAGAPPGGPLVLSTFPSTPLVTEQDGCGPSGAGASNVFVQMRTEVGPVKAAQAQTLVLTQAPLVWQAPGALCGGVVCPPPLLLAAAPVVPVMAA |
|---|
| 1 | 4dkmA | 0.09 | 0.07 | 2.66 | 0.61 | CEthreader | | --------------------------LPTTHEVHVYGSINGVEFDLVGSGKGNPKDGSEEIQVK-STKGPLGFSPYIVVPNIPDGMSPFQAAADDGSGYVVHRTIQFEDG-ASLTGNYRYSYDGGHIKGEFHVVGSGFPADG----------PVMTKSLTAVDWSVATMLFPNDTTVVSTIDWTCPTTS----GKRYHATLRTNYTFAKPIAATILQKQPMFVFRKTEVKAS-----------DAEINLKEWQKAFHDL- |
| 2 | 7jtkA3 | 0.07 | 0.05 | 1.94 | 0.58 | EigenThreader | | SRSSFNGDTYFGSYDVKHGPGLYAFATGAGYAGEYAGG--KRHGRGVMVF---------PDGGTYVGEF--VAD---KFEGQGQYRY----PDGSVYTGSWAAGQKHG----PGVYWDTARGLLV--------------------GYEQ------------------------PALRFE----GEFVR-------GMPAGTATYTLTGHRTLDHIQAEEGPTLALPCAYGIPPGSGDPPLPAEGLTFTAEQLPPDTVFPP |
| 3 | 6w09E | 0.16 | 0.15 | 5.09 | 0.33 | FFAS-3D | | -VYKAIRPYLAHCPDCGEGHSC---HSP-VALERIRNEATDGTLKIQVSLQIGIKTDDSHDWTKLRYMDN------HMPADAERARLFVRTSAPCTGTMGHFI-LARCPKGETLTVGFTDSGKISHSDPPVIGREKFHSRPQHRKELPCSTYAQSTAATAEEIEVHMPPDTPDRTLMVKITVNSQTVRYKCNCGDSNEGLTT----TDKVINNCKVDQCHAAVTNHKKVPRNAELGDRKGKVHIPFPLANVTCRVPKARN |
| 4 | 6ybt2 | 0.09 | 0.08 | 3.12 | 1.03 | SPARKS-K | | ILLQGHERSITQIKREGDLLTVAKDPIVNVWYSVTYMGHTGAVWCVDADWDTKGKQLATNSAVRTCGFDFIMFSTFVSFFDLRYMKIPCNDSKITSAVWGPLGECIIAGHESGELNQYSAKSGEVKEHSRQI-NDIQLSRDVTASKDNTAKLFDSTTLERTERPVNSAALSPNYDHVVLGGGQE---AMDVTTTSTRIGKFEARFFHLAFEEEFGRVKGHFGPINSVAFHPDGKSYSSGGEDGYVRIH------------ |
| 5 | 5xm3A | 0.09 | 0.02 | 0.73 | 0.47 | CNFpred | | ---------------------------------------------------------------------------------------------------------------------------------------------------------------------GWYAYDPQLDMFYYGSGNPAPWNETMRPGDNKWTMTIWGRDLETG--------LAKFGYQKTP-------------------------------- |
| 6 | 5awoA | 0.06 | 0.04 | 1.85 | 0.67 | DEthreader | | --NIYGIACTDISRTTNGYINWVHRDFFWLDVTAK-QV---RIDFLSWYEDGRQVNAPHGRNYELALWINEAAGEDMEVSLVPHMFQDGS------RINAD---------------------NWQDANWAPFCGFTGW----SHRNGILGSPLIAWYTNKEVLQLGVALFNRSDVTKTIDFAKLGPAAWAGFNNHPGYDGNG-------------------------VTFAV-RYRYAWGVANLNW-------------- |
| 7 | 3v4pA | 0.07 | 0.07 | 2.86 | 0.97 | MapAlign | | VLHSHGANRWLLVGAPTAIYRCRIGKNIAPCYQDYVKKFGENFASCQAGISSFYTKDLIVMGAPGSSYWTGSQVKFGSYLGYSVGAGHFRSQHTTEVVGGAPQHFSIDEKELNILFGASVCAVDLNADGFSDLLVGAPMQSTIREEGRVFVYINSGSGAVMNAMETNLVGSDKYAARF--GESIVNLGDIDNDGFEDVAIGAPQEYIYNGRADGISSTFSQRILSMFGQSISGQIDADNNGYVDVAVGAFRSDSAVLLR- |
| 8 | 5ay6A | 0.11 | 0.11 | 4.00 | 0.64 | MUSTER | | VGVNARSEQPV---AAAVSYKVGTAGSPSKTNVVDSATNSHNYDVVYSSTGIANPVSGNNEYLVDIKENGVIVATGKVAYDAATNELVSSTIDYKGASPVTGSMTTTRINAAGTTVNLADLGIVNASGADDAEVVAGKLYDPSTWSMSDYAKDNSKGVKPDFEVQIPLSDSKGGQMLKGPGPNQ-LRAKPGDLANNGNGQISTGIIEFTTDGKLKNTGSLFGTTSPTAITIKSSGYIAPTVTPPAVQPPTPPTWADALGI |
| 9 | 2pffB | 0.20 | 0.18 | 5.78 | 0.75 | HHsearch | | LELENPSNTPDKDYLLSIPISCPLGFTPGE-LRSYLKGATGHSVTAVAIAETDSWEVSVRKAITVLFFIGVRCYEAYPNTSLPSILEDSLENNEGVPS--PMLSISNLTQEQVQDYAGKQEISLVNGAKNLVGLNLTLRKA-------KAPSGL----DQSRIPFRKLKFSNRFLPVASPFHSHLLVPAS---------DLINKDLVKNNVS-FNAKDIQ--IPVYDTFDGSDLRVLSGSIDCIIRLPVKWETTTQFKAT |
| 10 | 4k3yA | 0.08 | 0.08 | 3.15 | 0.59 | CEthreader | | SSCHDGFQWTVLSVAGDGFVSILYGGIITDTIHPTNGGPLRICNDGTCYTIIADGTTYTASSHRLYRLWKALDTTGFNFEFPTCYYTSGKVK-------CTGTNLWNDAKRPFLEFDQSFTYTFKEPCLGFLGDTPRGIDTTNYCDKTTTEGEGGIQGFMIEGSNSWIGRIINPGSKKGFEIYKFLGTLFSVQTVGNRNYQLLSNSTIGRSGLYQPAYESRDCQELCFWIEIAATTKAGLSSNDLITFCGTGGSMPDVNW |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|