Methods
Starting from a query amino acid sequence, DeepMSA2 is used to search the query against
multiple whole-genome and metagenome sequence databases to create a multiple sequence
alignment (MSA). The MSA is then used by DeepPotential to derive input features based
on co-evolutionary analyses for the deep ResNet training.
DeepPotential outputs the probability distribution of Cβ-Cβ/Cα-Cα contact and
distance maps, as well as the inter-residue orientations.
These restraint potentials along with the inherent statistical energy function are
used to guide the L-BFGS folding simulations to final full-length structure model construction
(see Fig. 1).
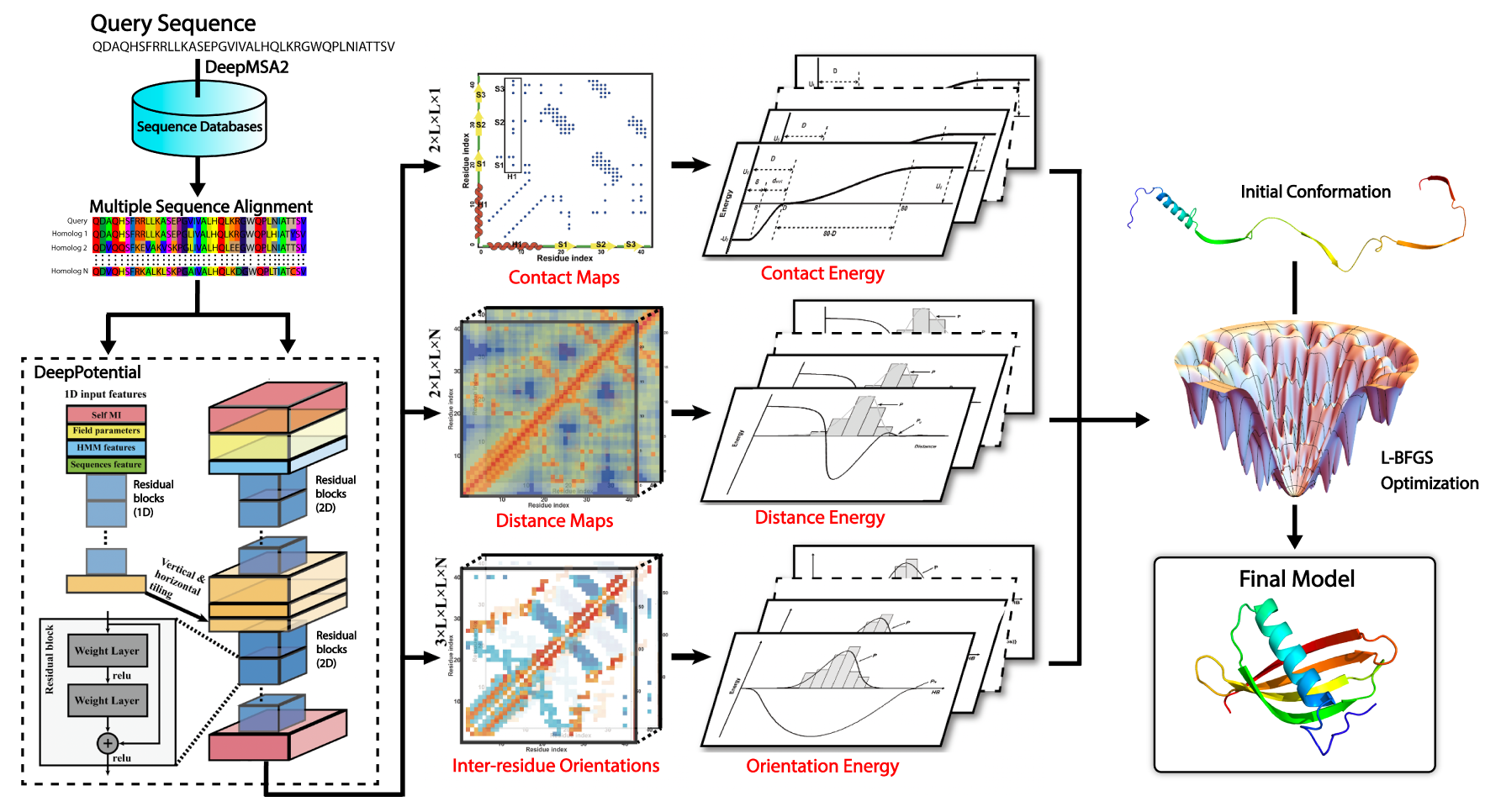 Figure 1. Pipeline of DeepFold for deep-learning based ab initio protein structure prediction.
Figure 1. Pipeline of DeepFold for deep-learning based ab initio protein structure prediction.
Assessment of DeepFold for protein structure prediction
DeepFold was tested on a set of 221 Hard threading targets collected from SCOPe and CASP9-12
as well as the CASP13 FM targets (Figure 2). The results show that the modeling
performance of DeepFold was significantly greater than other deep learning approaches
such as DMPfold and trRosetta, as well as the leading template-based modeling methods
such as I-TASSER. It also outperformed AlphaFold v1.0 on the CASP13 FM targets (see Fig. 2).
 Figure 2. Head-to-head TM-score comparisons between DeepFold and
other protein structure prediction methods:
A) I-TASSER; B) C-I-TASSER; C) DMPfold; D) trRosetta; E) AlphaFold.
(A-D) are based on the 221 Hard benchmark proteins,
while (E) is on 31 FM targets from CASP13.
Figure 2. Head-to-head TM-score comparisons between DeepFold and
other protein structure prediction methods:
A) I-TASSER; B) C-I-TASSER; C) DMPfold; D) trRosetta; E) AlphaFold.
(A-D) are based on the 221 Hard benchmark proteins,
while (E) is on 31 FM targets from CASP13.
Server inputs
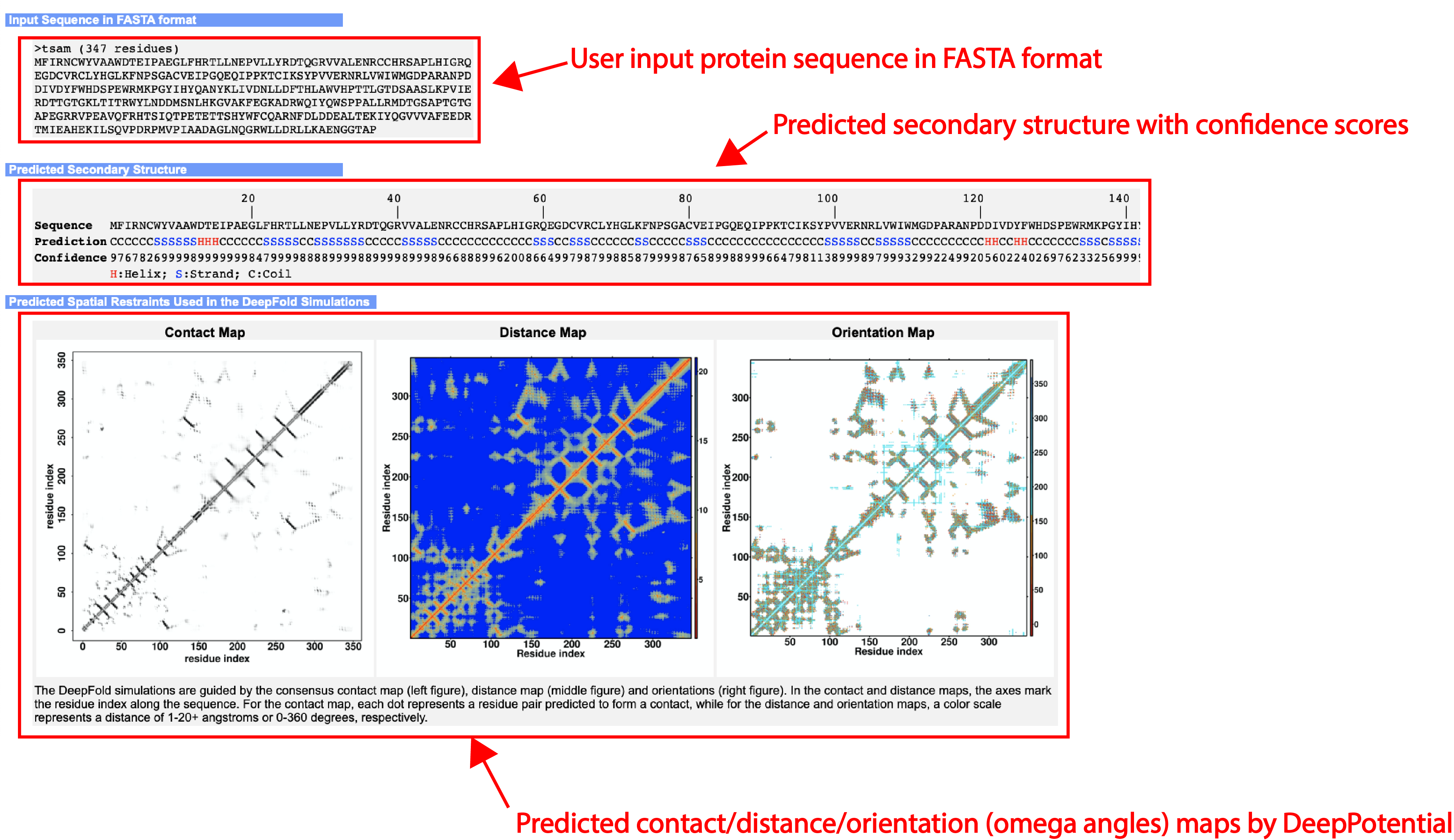
The user needs to paste the fasta-formatted amino acid sequence into the input box,
or upload the amino acid sequence of the query protein using the "Choose file" button (see Fig. 3).

Figure 3. Illustration of input for the DeepFold server.
Server outputs
The output of the DeepFold server include:
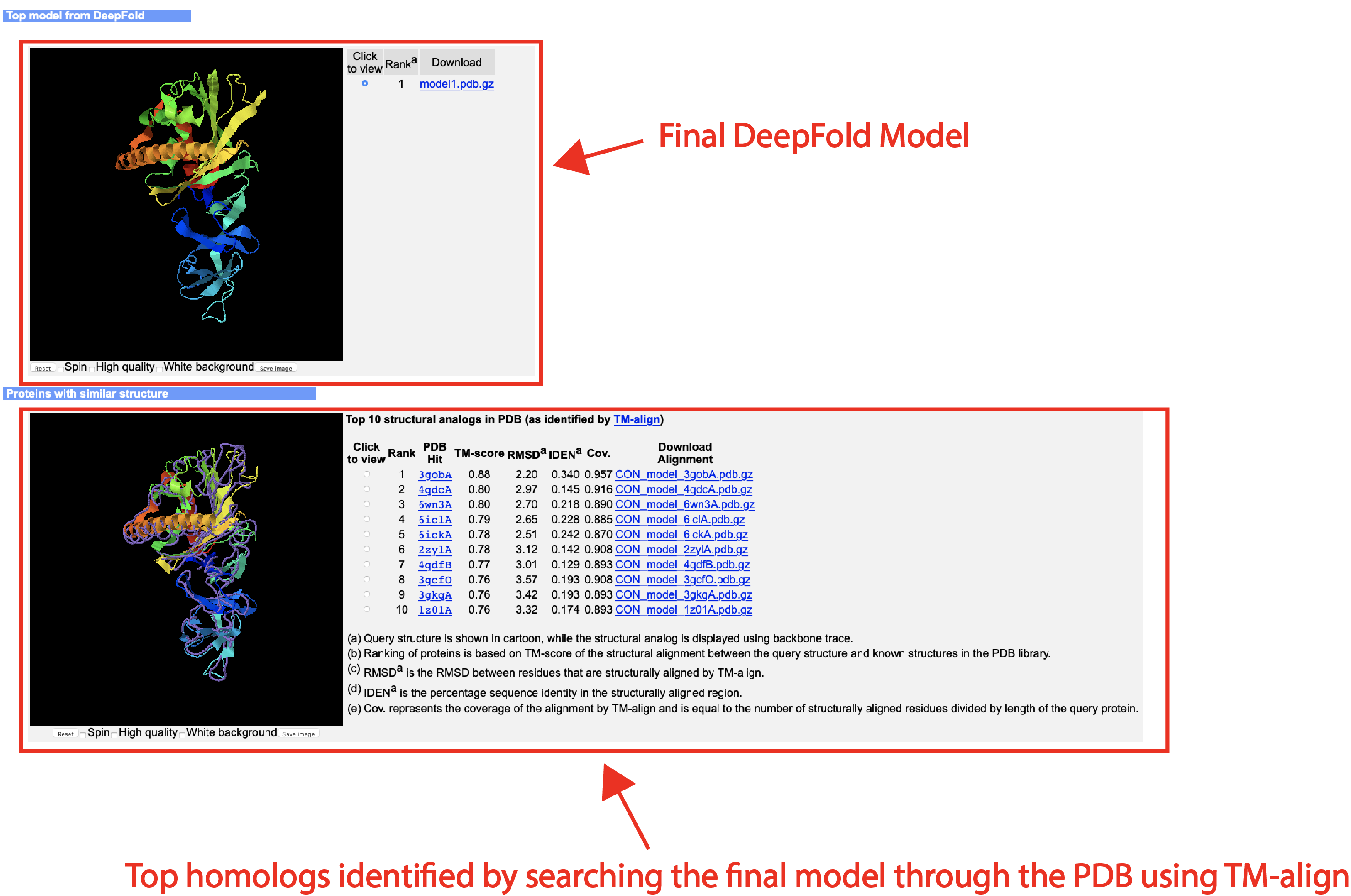
- The full-length atomic model
- Predicted secondary structures
- Predicted contact, distance, and orientation maps
- Top 10 proteins in the PDB that are structurally closest to the predicted models
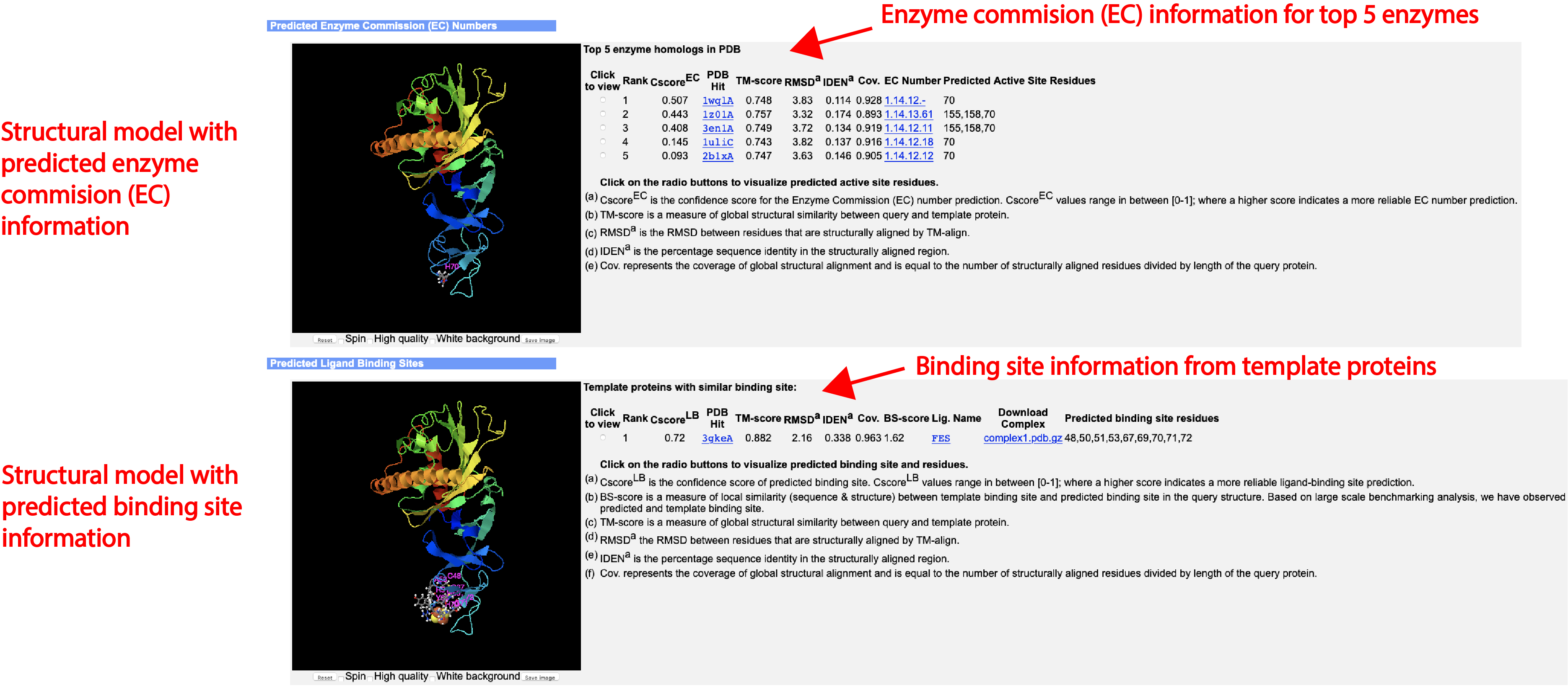
- Predicted Enzyme Classification and the confidence score (if you check the "Predict protein function based on structure model (running time may be doubled)." option of the input)
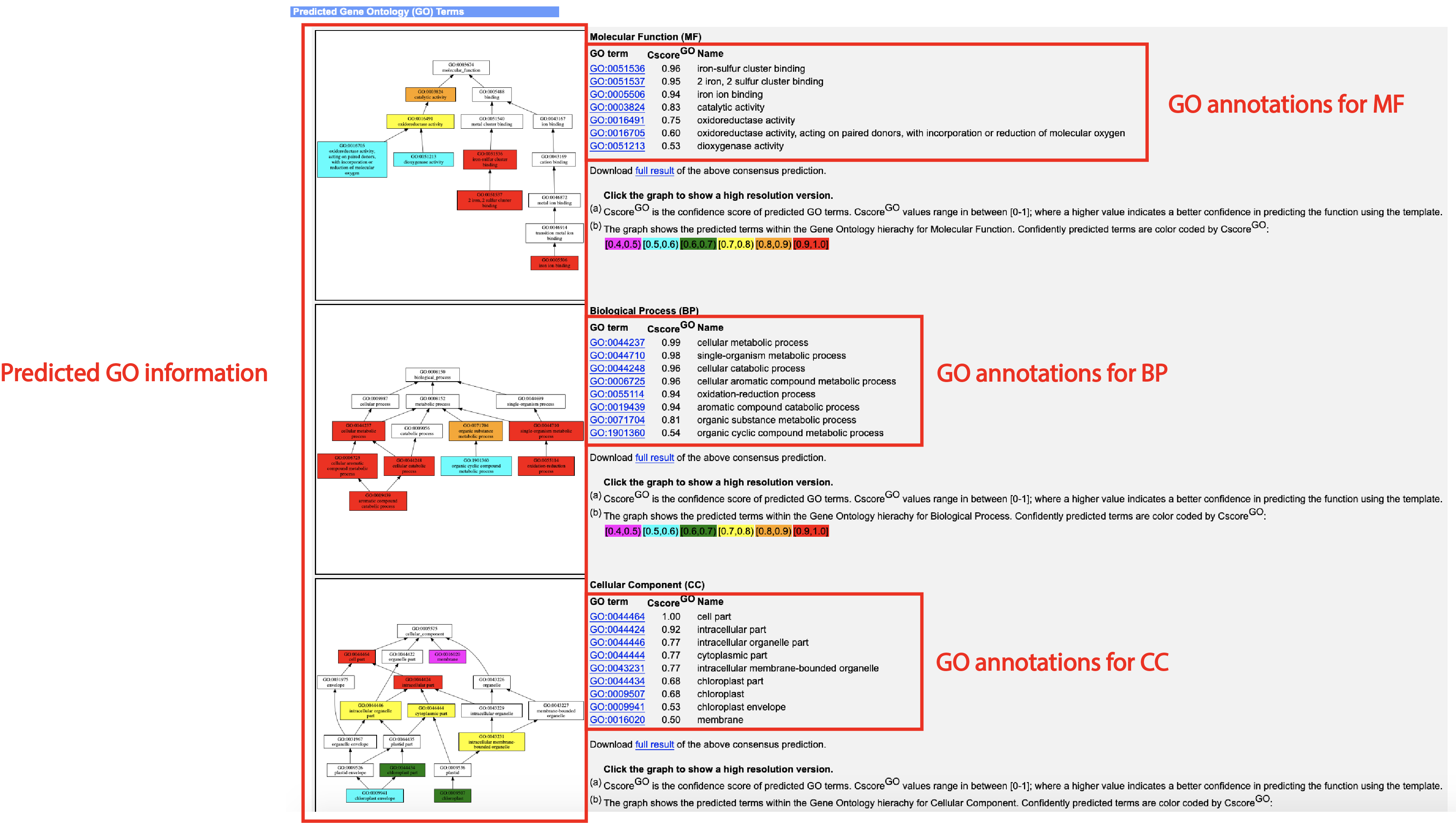
- Predicted GO terms and the confidence score (if you check the "Predict protein function based on structure model (running time may be doubled)." option of the input)
- Predicted ligand-binding sites and the confidence score (if you check the "Predict protein function based on structure model (running time may be doubled)." option of the input)
An illustrative example of the DeepFold output can be seen from below (Figs. 4-7):
How to cite DeepFold?
-
Robin Pearce, Yang Li, Gilbert S. Omenn, Yang Zhang.
Fast and Accurate Ab Initio Protein Structure Prediction Using Deep Learning Potentials
(submitted).
[back to the DeepFold server]