C-QUARK:
Understanding the result output.
The full modeling result for each job is available as a
tarball download at the top of output webpage. Here is the
description of format for each file in the tarball:
- seq.txt:
Input sequence in FASTA format.
- model1.pdb - model10.pdb:
Top 10 final structure model in PDB format. If the structure modelling has high
confidence, there might be less than 10 final models.
- seq.dat.ss:
Three-state secondary structure prediction in the following format:
position (starting from 1), amino acid type, predicted secondary structure type,
confidence as random coil, helix, β-strand.
- turn.txt:
β turn prediction in the following format:
position (starting from 0), no use, confidence value c (in [-1,1] and
predicted as a beta-turn when c>0).
- contact.txt:
Distance profile derived from fragments in the following format:
position i (starting from 0), position j (starting from 0), avg., std., number
of fragment pairs, 0.5Å, 1.0Å, 1.5Å, 2.0Å, 2.5Å,
3.0Å, 3.5Å, 4.0Å, 4.5Å, 5.0Å, 5.5Å,
6.0Å, 6.5Å, 7.0Å, 7.5Å, 8.0Å, 8.5Å,
9.0Å, peak position in array, number of fragment pairs in peak, atomic
distance for fragment pairs in peak.
- topdh.topdh:
Clustered torsion angle pairs from fragments in the following format:
position (starting from 0), cluster number.
amino acid, Cα x, Cα y, Cα z, secondary structure,
Cα φ, Cα-Cα, Cα-Cα-Cα, ψ, CN,
Cα-CN, ω, N-Cα, CN-Cα, φ, Cα-C, N-Cα-C,
position in template, template name, accumulated probability.
- phi.txt,
psi.txt:
Predicted φ and ψ backbone torsion angles in the following format:
position (starting from 1), predicted value t in [-180°,180°]
- sol.txt:
Predicted solvent accessibility in the following format:
positon, no use, predicted value s (in [-1,1])
- alldecoy.pdb:
All 5000 decoy structures from C-QUARK simulation in multi-model PDB format.
Only Cα is included.
- contact.map:
Selected residue contacts used as restraints for C-QUARK simulation.
This file is a consensus combination of in-house programs
(ResPRE,
NeBcon,
RICMAP,
DeepPLM,
DeepPRE2)
and third-party predictors
(DeepCov,
DeepContact,
DNCON2,
GREMLIN,
MetaPSICOV,
MetaPSICOV2).
- cscore.txt:
Confidence score (C-score) and estimated TM-score for C-QUARK models. Higher C-score and estimated TM-score usually indicate better model quality.
Note that, in general, structure quality estimation is only accurate for the first model.
The first model is on average the most reliable and should be considered if without special reasons (e.g., from biological common sense or experimental data).
|

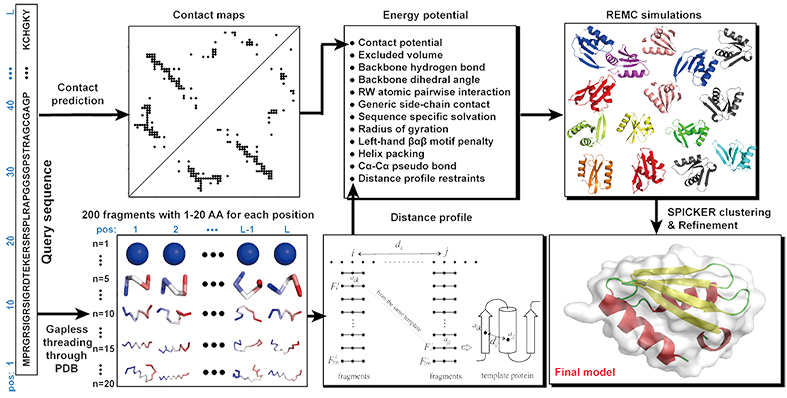 Figure 1. Pipeline of C-QUARK.
Figure 1. Pipeline of C-QUARK.